В настоящее время эпилепсия входит в пятерку наиболее распространенных неврологических заболеваний. Она нередко дебютирует уже в детском возрасте и относится к потенциально инвалидизирующим патологиям. Причем в большинстве случаев диагностируется идиопатическая форма эпилепсии. Это классический и наиболее часто встречающийся вариант заболевания, с несколькими клиническими разновидностями и вариантами течения.
Что это за болезнь

Эпилепсией называют хроническое заболевание, обусловленное патологией на уровне головного мозга и проявляющееся преимущественно приступами (припадками) судорожного и бессудорожного характера. Свойственны ей и другие, менее яркие симптомы. Изучением всех связанных с эпилептической болезнью проблем занимается особая наука эпилептология, но в повседневной клинической практике диагностику и лечение нередко проводит невролог (невропатолог).
Согласно действующей классификации, эпилепсия подразделяется на идиопатическую, симптоматическую и криптогенную. Эти неоднородные по происхождению и симптоматике состояния отличаются друг от друга характером изменений в центральной нервной системе.
Идиопатическая эпилепсия относится к самостоятельно возникающим заболеваниям. Раньше ее называли истинной, генуинной, первичной. Она характеризуется несколькими признаками:
- В головном мозге нет органических (структурных) изменений. Поэтому такой форме болезни не свойственны очерченная очаговая неврологическая симптоматика, задержка психомоторного развития или когнитивный регресс. Современные высокоточные методы нейровизуализации оказываются не информативными.
- Приводящие к припадкам нарушения носят функциональный характер и не обусловлены действием каких-либо внешних факторов или появившихся в течение жизни заболеваний.
- Склонность к раннему проявлению симптоматики. Идиопатическая эпилепсия в 70-75% случаев дебютирует в детском возрасте, с основным пиком заболеваемости у подростков в 9-14 лет. Поэтому обычно речь идет о детских и ювенильных (юношеских) формах болезни.
- Наследственная предрасположенность. В настоящее время выделено около 500 генов в соматических хромосомах, мутация в которых может стать причиной эпилепсии. Многие формы болезни характеризуется аутосомно-доминантным или рецессивным типом наследования.
- Достаточно высокая чувствительность к современным противосудорожным препаратам. Это позволяет в большинстве случаев добиться значительного урежения приступов или даже перевести заболевание в стойкую ремиссию.
На долю идиопатических типов приходится около 70-75% случаев эпилепсии с различными клиническими проявлениями, при этом преобладают ее генерализованные формы.
Что происходит в мозге при идиопатической эпилепсии
Болезнь не связана с локальной гибелью или ишемией (выраженным кислородным голоданием) нервных клеток, опухолевым ростом, внутричерепной гипертензией, пороками развития или какими-либо другими патологическими процессами. Все изменения носят функциональный характер и формируются на клеточном и биохимическом уровнях.
Согласно современным представлениям, значительная часть случаев идиопатической эпилепсии обусловлена каналопатией – дисфункцией ионных каналов в стенках нервных клеток. Такая проблема обычно обусловлена патологией одного или нескольких генов, кодирующих структуру трансмембранных белков.
Результатом подобных каналопатий становится дефект транспорта отдельных ионов (калия, натрия) или целых молекул нейромедиаторов (ГАМК, ацетилхолина и др.), что приводит к комплексу нарушений:
- изменение скорости проведения сигналов в нервной ткани, склонность к каскадному распространению пароксизмальных импульсов;
- поддержание дисбаланса между процессами торможения и возбуждения в головном мозге, с преобладанием активирующих влияний;
- создание предпосылок для чрезмерной стойкой деполяризации на поверхности нейронов (она может быть запущена выраженным ионным дисбалансом или избыточным количеством глутамата, причем в подавляющем большинстве случаев без явного провоцирующего фактора).
При идиопатической эпилепсии соседние нейроны очень легко синхронизируются друг с другом, что объясняет склонность к быстрому тотальному распространению пароксизмально возникшего гипервозбуждения. В этот процесс генерализации вовлекаются и подкорковые регулирующие структуры, что ускоряет передачу эпилептического импульса. В результате возбуждение охватывает практически весь головной мозг, провоцируя приступ с различной симптоматикой.
При этом у каждого пациента формируется собственная достаточно стабильная схема вовлечения и реакции различных мозговых структур. Это определяет характерную клиническую картину болезни, с преобладанием определенных симптомов во время приступа.
Основные проявления
Ключевое проявление эпилепсии – разнообразные приступы, которые могут комбинироваться друг с другом или существовать в изолированном виде. Они бывают судорожными (сопровождающимися неконтролируемой пароксизмальной активностью скелетных мышц) и бессудорожными, фокальными и генерализованными.
Нередко развитию приступа предшествует аура. А при серии следующих друг за другом припадков с генерализацией говорят о развитии эпилептического статуса, что чревато развитием отека головного мозга.
При идиопатической эпилепсии возможно появление приступов любого типа. Но преобладание одного из них или характерная трансформация разных видов друг в друга позволяет дифференцировать формы болезни.
К основным вариантам припадков относят:
- Генерализованные классические «большие» тонико-клонические приступы и их варианты, с преобладанием одной из фаз.
- Абсансы, которые тоже относятся к генерализованным приступам. Они могут быть простыми с вегетативными нарушениями и замираниями, либо сложными (с автоматизмами различного характера, орофасциальными миоклониями, атонией, закатываниями глаз и др.).
- Парциальные или фокальные припадки, раньше их называли «малыми». Бывают моторного, висцеровегетативного, чувствительного типа, с психическими эпилептическими автоматизмами, нарушениями восприятия и ощущения схемы тела и др.
Для большинства идиопатических форм болезни не свойственно развитие неврологического дефицита и выпадение высших корковых функций. Физическое и интеллектуальное развитие у детей обычно не страдает, если нет осложняющих ситуацию сопутствующих заболеваний и предшествующих перинатальных вредностей.
Изменения личности по эпилептическому типу возникают нечасто, лишь при тяжело протекающих формах болезни с частыми плохо контролируемыми генерализованными приступами и эпистатусами.
Классификация
В настоящее время общепризнанной считается классификация, основы которой были приняты еще в 1981 году Международной Лигой по борьбе с эпилепсией (Киото, Япония). При этом учитывается характер имеющихся у пациента приступов, с оценкой их разновидности и склонности к генерализации.
В соответствии с таким подходом, все варианты болезни подразделяются на 2 основные группы: генерализованные и парциальные (фокальные). Внутри каждой их них имеется несколько различных заболеваний, отличающихся по клинической картине, времени проявления симптомов и прогнозу.
Генерализованные формы
Выделяют несколько основных клинических разновидностей (по действующей классификации ILAE от 1989 г.):
- Детская абсансная эпилепсия – бессудорожный вариант заболевания с доказанной наследственной природой. Характеризуется частыми многократными приступами по типу абсанса у детей 4-9 лет. На нее приходится 10-17% от всех идиопатических форм.
- Ювенильная (юношеская) абсансная эпилепсия, с дебютом в подростковом возрасте. Диагностируется примерно в 12% случаев, у ⅔ пациентов дебютирует в возрасте 9-13 лет. При этом первоначально у ребенка могут возникать и типичные генерализованные судорожные приступы, а в последующем преобладают абсансы.
- Доброкачественная младенческая миоклонус-эпилепсия. Дебютирует в возрасте от 4 мес. до 3 лет с миоклонических вздрагиваний, в последующем эти приступы приобретают генерализованный характер. Чаще заболевают мальчики.
- Ювенильная миоклонус-эпилепсия или синдром Янца. Распространенность колеблется от 4 до 12%. Патология связана с дефектом генов на коротком плече 6 соматической хромосомы. В клинике преобладают миоклонические припадки, хотя они могут дополняться эпизодически возникающими абсансами и генерализованными судорожными приступами.
- Идиопатическая эпилепсия с генерализованными судорожными приступами (изолированными).
- Эпилепсия с генерализованными судорожными приступами периода пробуждения. Заболевание с доказанной генетической детерминированностью и дебютом преимущественно в 9–11-летнем возрасте. Характеризуется появлением типичных генерализованных припадков с тонико-клоническими судорогами в просоночном состоянии.
- Эпилепсия с фотосенсибилизацией. Характеризуется рефлекторным возникновением приступов в ответ на зрительную стимуляцию, с преобладанием генерализованных судорожных тонико-клонических припадков. Пик заболеваемости приходится на возраст 12-14 лет, но первичные проявления возможны вплоть до 25-летнего возраста.
Генерализованная идиопатическая эпилепсия – самый частый вариант заболевания, именно она определяет почти ⅔ случаев эпиприпадков в детском и подростковом возрасте.
По теме: Эпилепсия у детей
Фокальные формы
Эта полиморфная группа включает несколько заболеваний.
- Доброкачественные семейные судороги у новорожденных или доброкачественная семейная неонатальная эпилепсия
Один из прогностически благоприятных вариантов заболевания, несмотря на очень ранний дебют симптоматики. Первые проявления отмечаются обычно уже на 1-й неделе жизни ребенка, в виде фокальных коротких приступов с апноэ. В последующем могут присоединяться двигательные автоматизмы или клонические подергивания. Симптоматика обычно угасает к началу 2-го года жизни. Заболевание относится к редким и имеет аутосомно-доминантный тип наследования.
- Идиопатическая парциальная эпилепсия с лобными автоматизмами
Дает о себе знать обычно в возрасте 2-8 лет. Характерна дифференцировка симптоматики в ночное и дневное время. В состоянии бодрствования у ребенка преобладают сложные парциальные и абсансоподобные приступы, а во сне появляются гемифациальные (с вовлечением половины лица) моторные припадки, которые иногда имеют склонность к вторичной генерализации. Активный период болезни длится несколько лет, после чего обычно наступает стойкая ремиссия.
- Затылочная доброкачественная эпилепсия с поздним началом (синдром Гасто)
Пик заболеваемости в 8-9 лет. Проявляется непродолжительными сложными по структуре фокальными приступами: начало припадка со зрительных галлюцинаций, которые сменяются кратковременным исчезновением зрения (корковой слепотой) и односторонними клоническими судорогами. Выход из приступа через мигренозные боли.
- Семейная височная эпилепсия
Характеризуется простыми или сложными парциальными припадками и предшествующей психосенсорной аурой. Один из вариантов болезни – приступы в виде слуховых галлюцинаций, иногда дополняющиеся вегетативными расстройствами. Дебютирует обычно в подростковом возрасте, но нередко первые ее проявления возникают лишь к 25-30 годам. Очень хорошо поддается медикаментозной терапии и не склонна к трансформации или генерализации.
В последние годы некоторыми исследователями выделяется еще одна особая форма болезни: идиопатическая фокальная эпилепсия с псевдогенерализованными приступами. У пациентов с таким диагнозом изначально возникающие фокальные приступы быстро развертываются и усложняются, что клинически выглядит как их генерализация. Но при этом ЭЭГ однозначно показывает, что происходит лишь вторичная билатеральная синхронизация, без тотального охвата всего мозга.
Редкие клинические формы
К редким формам заболевания относится синдром Панайотопулоса или идиопатическая затылочная эпилепсия с ранним началом, дебютирующая в возрасте от 1 до 13 лет. Характеризуется тяжело протекающими приступами с ротацией (поворотом) глаз, выраженным вегетативным компонентом и длительно сохраняющимся бессознательным состоянием. Чаще всего они начинаются в период сна, и нередко первым проявлением припадка становится не приносящая облегчения рвота.
Риск развития эпилептического статуса при синдроме Панайотопулоса очень высокий. Но заболевание в целом оценивается как прогностически благоприятное, с доброкачественным течением и сохранностью у ребенка интеллектуально-мнестических функций. В большинстве случаев приступы возникают редко, всего 1-3 раза в течение жизни, не оказывают негативного влияния на развитие ребенка и на общее состояние его здоровья.
Диагностика
Обследование включает консультацию невролога, функциональную диагностику (ЭЭГ, сомнография, суточное мониторирование) и современные методы нейровизуализации для исключения органической причины припадков.
Чаще всего для исследования головного мозга назначают МРТ или КТ, предпочтительно с ангиопрограммой и дополнительным контрастным усилением. Это позволяет исключить объемные образования, пороки развития, выраженные нарушения ликвородинамики, сосудистые изменения (в том числе врожденные мальформации) и другую патологию, способную привести к очаговой эпилептической активности. Такое обследование необходимо для дифференциальной диагностики и подтверждения идиопатической природы заболевания.
Информативность ЭЭГ может быть различной. Это зависит от периода, когда проводилось исследование, формы и тяжести течения болезни. В приступный период ЭЭГ выявляет генерализованную синхронизированную билатеральную (двустороннюю, с охватом обоих полушарий головного мозга) эпилептическую активность по типу пик-волна. Разряды короткие, обычно провоцируются гипервентиляцией и фотостимуляцией, вероятность их выявления повышается при депривации (целенаправленном лишении) сна.
В большинстве случаев в межприступную (интериктальную) фазу ЭЭГ не выявляет полипиковой и генерализованной судорожной активности, общий (основной) ритм сохранен. Функциональные признаки структурных дефектов не характерны. Исключение составляет ювенильная миоклонус-эпилепсия, при которой аномальная ЭЭГ выявляется практически постоянно у 80-95% пациентов.
При некоторых формах заболевания могут выявляться также другие аномалии: доминирующий окципитальный (затылочный) дельта-ритм, эпизодическое бифронтальное (в лобных отделах обоих полушарий) замедление ритма и др.
Как лечить
Пациентам с любыми эпилептическими припадками показана диспансеризация с регулярным контролем состояния. Лечение подбирается индивидуально неврологом или эпилептологом. Иногда требуется также участие психиатра, если клиническая картина включает психовегетативные пароксизмы, обманы восприятия (галлюцинации), нарушения поведения и когнитивные расстройства.
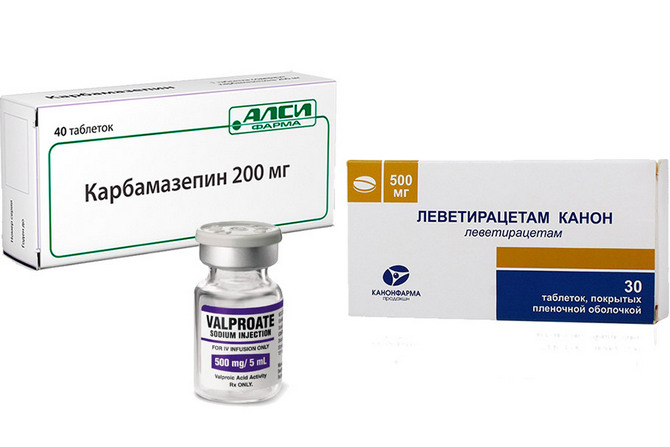
Основой лечения является медикаментозная терапия, с использованием антиконвульсантов (противосудорожных средств) различных групп. К наиболее употребимым препаратам относят:
- препараты вальпроевой кислоты (вальпроаты);
- Леветирацетам (Топирамат);
- Карбамазепин.
Препараты и схема их приема подбираются индивидуально, с учетом вида возникающих у пациента припадков. Лечение начинают с назначения малых доз средства 1 линии, с последующим подбором дозировки. При его недостаточной эффективности принимается решение о смене препарата или переходе на комбинированную терапию.
Противосудорожные средства принимаются постоянно, при этом необходимо регулярно контролировать картину крови и биохимические показатели работы печени. Продолжать лечение рекомендуется в течение не менее 5 лет после последнего приступа, отмена препаратов проводится постепенно, под контролем врача.
У 3-5% пациентов встречается медикаментозная резистентность – устойчивость к применяемым лекарствам, с отсутствием положительной динамики даже на комбинированной терапии достаточными дозами. В этом случае при некоторых формах заболевания может быть принято решение о целесообразности хирургического лечения, с проведением резективных или функциональных операций на головном мозге. Иногда проводят также стимуляцию блуждающего нерва.
По теме: Таблетки от эпилепсии
К немедикаментозным методикам относят соблюдение режима дня, исключение провоцирующих (стимулирующих) факторов и диету. Желательно избегать потребления кофе, крепкого чая, чрезмерно острой пищи, алкоголя, тонизирующих напитков. Иногда врач дает рекомендации о переводе пациента на кетогенную диету – особую систему питания, когда ведущим источником энергии становятся жиры, а углеводы в рационе строго ограничиваются.
Прогноз
В целом идиопатические формы эпилепсии имеют относительно благоприятный прогноз. Многие из них дебютируют в детско-подростковом возрасте и в последующем постепенно угасают, иногда давая о себе знать лишь при явных провоцирующих факторах у взрослых.
Исключение составляют некоторые тяжело протекающие формы эпилепсии. Резистентность (устойчивость) к терапии нередко отмечается также у абсансных форм болезни, что требует тщательного подбора комбинации препаратов. Но даже в этом случае у ⅓ пациентов удается достичь лишь улучшения в виде значительного урежения и упрощения абсансов и исчезновения дополняющих их судорожных припадков.
Возможна также трансформация форм болезни друг в друга, несмотря на изначальное поражение разных генов. Например, эпилепсия с генерализованными приступами пробуждения может перейти в ювенильные формы абсансной или миоклонической эпилепсии. А у 30% девочек она в последующем трансформируется в катамениальную эпилепсию, при которой первично-генерализованные судорожные приступы возникают в предменструальный период.
Но в большинстве случаев удается добиться хорошего медикаментозного контроля над частотой и выраженностью приступов, избежать социально-бытовой дезадаптации пациентов и сохранить хороший темп интеллектуального развития ребенка. В стойкую ремиссию уходят 65-85% пациентов, причем большинству из них во взрослом возрасте уже не требуется постоянная поддерживающая терапия.